
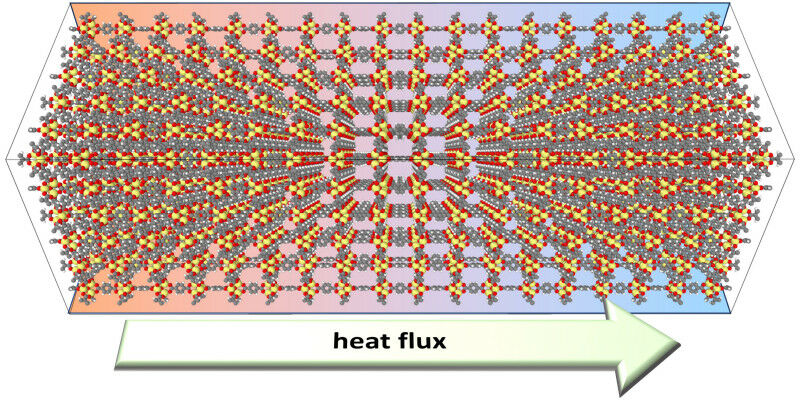
Devido às estruturas complexas de cristais microporosos conhecidos como MOFs, simulações confiáveis de suas propriedades têm sido difíceis até agora. O aprendizado de máquina fornece a solução.
Armazenamento de hidrogênio, condução de calor, armazenamento de gás, CO2 e sequestro de água – as estruturas metal-orgânicas (MOFs) possuem propriedades extraordinárias devido à sua estrutura única na forma de cristais microporosos, que possuem uma área superficial muito grande, apesar de seu pequeno tamanho. Isso os torna extremamente interessantes para pesquisas e aplicações práticas. No entanto, os MOFs são sistemas muito complexos que até agora exigiram muito tempo e poder computacional para serem simulados com precisão. Uma equipe liderada por Egbert Zojer, do Instituto de Física do Estado Sólido da Universidade de Tecnologia de Graz (TU Graz), melhorou significativamente essas simulações usando aprendizado de máquina, o que acelera bastante o desenvolvimento e a aplicação de novos MOFs. Os pesquisadores publicaram seu método na revista Nature Research npj Computational Materials.
Anteriormente irrealista para simular com a precisão dos métodos da mecânica quântica
“Para simular certas propriedades de MOFs, é necessário simular supercélulas enormes. Isto se aplica, por exemplo, ao cálculo da condução de calor em MOFs, o que é altamente relevante para quase todas as aplicações. As supercélulas simuladas geralmente contêm dezenas de milhares ou mesmo centenas de milhares de átomos. Para estes sistemas enormes, é então necessário resolver as equações de movimento de cinco a dez milhões de vezes. Isto está muito além das possibilidades computacionais atuais usando métodos confiáveis de mecânica quântica”, diz Egbert Zojer, descrevendo o desafio que precisava ser resolvido.
Assim, até agora, campos de força transferíveis, muitas vezes parametrizados com base em experiências, eram frequentemente utilizados para tais cálculos. No entanto, os resultados obtidos com tais campos de força revelaram-se geralmente não suficientemente fiáveis. Isto agora é fundamentalmente alterado pelo uso de potenciais aprendidos por máquina. Estes são adaptados a simulações de mecânica quântica, utilizando uma interação recentemente desenvolvida de algoritmos existentes, incluindo abordagens desenvolvidas na Universidade de Viena. Para o necessário aprendizado de máquina específico do material dos potenciais, as simulações da mecânica quântica precisam ser realizadas apenas para estruturas comparativamente poucas e significativamente menores. Como resultado, os cálculos são executados muito mais rapidamente e é possível simular as forças nas enormes supercélulas muitos milhões de vezes em supercomputadores modernos. A vantagem decisiva aqui é que não há perda relevante de precisão em comparação com a realização de simulações usando métodos de mecânica quântica.
Pesquisa mais eficiente dos imóveis desejados
Para o exemplo da condução de calor de MOFsisto significa que a estratégia de simulação recentemente desenvolvida tornará possível simular as propriedades relevantes do material mesmo antes do MOFs são sintetizados, permitindo assim desenvolver estruturas personalizadas no computador de forma confiável. Isto representa um grande avanço na investigação de materiais complexos, que para o transporte de calor permitirão, por exemplo, aos investigadores optimizar a interacção entre os nós de óxido metálico e os ligantes orgânicos semicondutores. O uso da nova estratégia de simulação também tornará mais fácil superar desafios complexos. Por exemplo, MOFs devem ter boa ou má condutividade térmica dependendo da sua aplicação.

Um sistema de armazenamento de hidrogênio, por exemplo, deve ser capaz de dissipar bem o calor, enquanto em aplicações termoelétricas uma boa condução elétrica deve ser combinada com a menor dissipação de calor possível. Além de simular a condutividade térmica, os novos potenciais aprendidos por máquina também são ideais para calcular outras propriedades dinâmicas e estruturais de MOFs. Estes incluem estruturas cristalográficas, constantes elásticas, bem como espectros vibracionais e fônons, que desempenham um papel decisivo na estabilidade térmica de MOFs e suas propriedades de transporte de carga.
Números quantitativamente confiáveis
“Temos agora ferramentas que sabemos serem incrivelmente eficientes para nos fornecer números quantitativos fiáveis. Isto permite-nos alterar sistematicamente as estruturas do MOFs nas simulações, sabendo ao mesmo tempo que as propriedades simuladas serão precisas. Isto nos permitirá, com base na causalidade, compreender quais mudanças na estrutura atomística geram os efeitos desejados”, diz Egbert Zojer, que sabe, através de grupos de pesquisa em Munique e Bayreuth, que já adotaram a nova estratégia de simulação, apesar de sua recente publicação. .
Esta área de investigação está ancorada na Área de Atuação” Ciência Avançada de Materiais “, um dos cinco focos estratégicos da TU Graz.
<